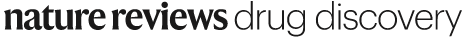
研究介绍
•
摘要介绍
Abstract
随着科学技术的飞速发展,药物开发领域虽取得显著进步,但高淘汰率的问题依旧严峻。面对这一挑战,作者提出深入分析药物候选物的药理学特征至关重要,特别是针对G蛋白偶联受体(GPCRs)这一庞大而复杂的受体家族。GPCRs的结构变换,如偏向性信号和变构调节,对药物作用机制具有深远影响。为了更精准地理解这些复杂机制,新检测方法和结构生物学的突破,如冷冻电子显微镜(冷冻电镜)技术,为科学家们提供了前所未有的分析工具。这些进步不仅加深了我们对药物与GPCRs相互作用的理解,也为新药开发提供了更为坚实的科学基础,从而有望显著提高新药开发的成功率。
引言介绍
Introduction
在小分子药物发现领域,筛选技术的进步已经为药物研发带来了革命性的变化。细胞检测、计算机筛选和生物物理方法等现代技术的引入,极大地提高了筛选命中率,使得以前难以攻克的靶点药物候选物的识别成为可能。这些技术不仅加速了药物开发的流程,也为新药研发带来了更多可能性。
然而,尽管筛选技术取得了显著进步,药物候选物在临床试验中的高失败率仍然是一个不容忽视的问题。其中,疗效不足是导致失败的主要原因之一。深入分析后,可以发现疗效不足主要源于四个方面:一是 对治疗疾病所需的药理效应理解不足 ;二是 选择了不合适的靶点以期望实现预期治疗效果 ;三是 选择的药物候选物无法有效调控靶点 ;四是 未能全面表征药物候选物的效应,并据此在适当的患者群体中评估药物 。
本文将重点关注第三和第四个原因,特别是在小分子药物发现项目中,聚焦于G蛋白偶联受体(GPCRs)这一重要靶点。作者认为, 通过利用新的技术进步,如冷冻电子显微镜(cryo-EM)以及对偏向性信号和变构调节的深入理解,可以更好地表征和改造GPCR的行为,从而提高药物开发的成功率。
GPCRs作为疾病过程中重要的靶点,其激活或阻断通常能带来显著的治疗益处。然而,GPCRs通过多种构象状态来介导复杂的信号传导,涉及各种细胞内转导子。随着技术的进步,新型的功能检测方法使得新型分子的检测和表征成为可能,如偏向性激动剂和变构调节剂(图1)。这些分子能够优先激活某些信号通路或通过与受体特定部位结合来影响GPCR的行为,为药物开发提供了新的方向和策略。
因此,本文旨在探讨如何通过深入理解GPCRs的行为,结合新的检测技术,提高药物筛选的命中率,并开发具有新特性的药物候选物,以期在药物研发领域取得突破性的进展。
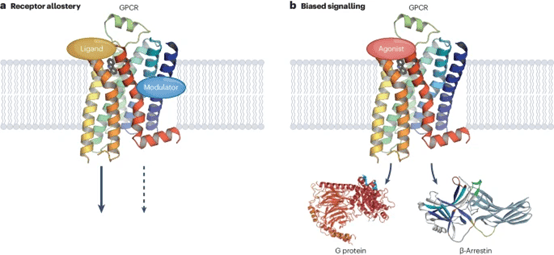
图1 以GPCR变构调节为靶标的小分子药物模型
正文
•
研究体外药理学特征分析点
在药物研发过程中,体外药理学特征分析是将初步筛选的化合物进一步优化为先导化合物乃至药物候选物的核心步骤。这一过程不仅关注化合物对治疗靶点的直接作用,还深入考量其安全性及ADME(吸收、分布、代谢和排泄)特性。药理学检测的设计,对于识别和优化这些特性至关重要。综合性检测方法在体外药理学特征分析中扮演着重要角色。与单一相互作用的检测相比,综合方法能够更全面地捕捉候选物的多种效应,为后续的优化工作提供更全面的指导(图2)。
针对GPCR(G蛋白偶联受体)靶向化合物的优化,特别注重结合模式、亲和力、效力和相互作用动力学等关键特征。这些特征的优化不仅关乎药物候选物与靶点之间的相互作用强度,还涉及到药物在体内的作用机制和效果。因此,通过精细调控这些特征,可以显著提高药物候选物的开发成功率,为临床治疗提供更有效、更安全的药物选择。
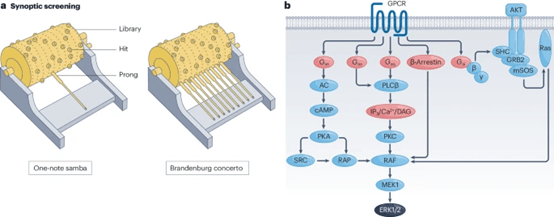
图2 药物筛选的综合筛查模型
结合模式
Binding mode
了解化合物与靶标之间的结合模式,特别是它是通过正构还是变构方式与靶标结合,对于揭示其作用机制至关重要。传统的药物发现过程中,大规模化合物库的筛选一直是检测变构调节剂的主要手段。
然而,随着科学技术的飞速发展, 近年来, 冷冻电子显微镜(cryo-EM)研究所提供的结构数据已成功应用于变构药理学的发现与研究之中。
亲和力
Affinity
在药物研发中,药物的亲和力是评估其与靶标受体结合能力的重要指标。药物的亲和力通常用Kd(平衡解离常数)的倒数来表示,反映了药物在特定浓度下与受体结合的能力。Kd值是药物与受体复合物解离速率(koff)与结合速率(kon)的比值,当药物浓度等于Kd时,大约50%的受体被药物占据。对于拮抗剂而言,Kd值是评估其影响体内信号传导能力的重要参数,但需要注意的是,Kd值可能会受到局部药物浓度的影响而有所偏差。对于激动剂来说,Kd值可能因测量系统的不同而有所变化,因为高亲和力和效力的激动剂在高受体表达或限制性微环境中可能会低估其真实效力。
此外,变构调节剂的亲和力评估更为复杂。特别是正变构调节剂(PAMs),由于它们与天然激动剂存在协同作用,其有效亲和力会受到体内天然激动剂浓度的影响。因此,在评估变构调节剂的亲和力时,需要综合考虑其与其他生物分子的相互作用。
效力
Efficacy
在药理学中,效力是指药物与靶标结合后所观察到的药理作用。这涉及配体所展现的多种可能效力以及它们对不同信号通路的偏向性。候选药物的总体治疗效力可能比我们最初认为的要复杂得多。虽然发现计划的主要目标是提供具有治疗价值的药物实体,但候选药物的效力通常仅从已知靶标的特性中推断。
特别是当靶标的信号能力具有多效性(即能够激活多种信号通路)时,激动剂的全部作用可能并不完全清楚。过去,药物效力往往被简单地视为细胞输出强度的单调梯度,但随着信号检测技术的不断发展,我们已经揭示了单个GPCR可以连接多个下游信号通路的复杂多样性。
通过使用多种检测手段,我们可以发现配体介导的多种效力。例如,检测单独的受体活性状态、检测不同G蛋白与受体的相互作用、以及通过NMR和受体结构识别不同受体活性状态等技术,可以帮助我们区分不同效力的分子。
选择性效力是由激动剂选择性稳定独特受体活性状态所引起的。这种选择性可以通过实验进行检查,如使用生物传感器检测单独的受体活性状态。不同的激动剂可能激活不同的受体状态,从而产生不同的信号特征,这对于药物研发和个性化治疗具有重要意义。
与靶点互作的动力学
Kinetics of interaction with the target
药物与靶点相互作用的动力学特性对于药物的治疗效果和选择性具有决定性作用。在体内,药物浓度是不断变化的,因此药物-靶点复合物的寿命成为一个关键参数。
在体外研究中,药物-靶点驻留时间模型被广泛用于优化药物动力学。尽管结合常数(kon)和解离常数(koff)共同决定了药物-靶点复合物的寿命,但通常koff在预测体内药代动力学特性时更为重要。这是因为koff直接反映了药物与靶点解离的速度,而药物-靶点驻留时间(1/koff)则可以通过体外测定来预测药物在体内的持续作用时间(图3)。
药物的治疗效果有时更依赖于药物-靶点复合物在体内的寿命,而非其效力。例如,某些抑制剂对特定病菌的疗效与药物-靶点复合物的驻留时间密切相关。对于血管紧张素II亚型1受体拮抗剂,结合时间和疗效之间也呈现出显著的相关性。
在某些情况下,药物在血浆中的半衰期可能较短,但其药效持续时间却很长。这通常是由于药物-靶点复合物的长驻留时间所致。例如,COMT抑制剂奥卡帕酮在血浆中的半衰期仅为1-1.4小时,但其药效持续时间却超过100小时,这正是由于它与靶点具有较长的驻留时间。
koff在比较药物对主要和次要靶点占用时间时也具有重要意义。对于某些时间敏感的反应,如神经效应,慢速的koff可能导致一种非竞争性拮抗现象。此外,新的测量工具显示,kon在某些情况下也是药物-靶点复合物寿命的重要指标,因为它反映了药物与靶点结合的速度。因此,在药物研发过程中,需要综合考虑kon和koff两个参数,以全面评估药物与靶点相互作用的动力学特性。
图3 体内药物代谢动力学以及药效学解离
体内活性转化
一旦确定了可进一步优化的前导化合物,验证该化合物在体内与靶标的真实作用就变得至关重要。这一验证过程涉及多个关键步骤和复杂的评估方法,以确保候选药物在体内的实际表现符合预期。
靶标区域内的存留情况
在评估药物在体内的药代动力学时,必须详尽考察药物的吸收与分布情况,以验证其是否能够有效地抵达目标区域。这一过程中需要解答几个关键问题:首先,要明确给药剂量中有多少药物真正被身体吸收;其次,需探究被吸收的药物具体分布到了身体的哪些部位;再次,要测定药物在体内的持续停留时间,即其清除率;最后,要计算达到血浆稳态浓度所需的给药频率。为了更准确地预测这些关键的药代动力学参数,可以利用成本相对较低的体外ADME(吸收、分布、代谢和排泄)试验进行模拟和预测。这种预测有助于提前了解药物在体内的行为模式,并有效减少临床试验中的潜在风险,为药物研发提供更为可靠的依据。
与体内靶标结合滞留时间
药物在治疗过程中要发挥效果,必须与治疗靶标有效结合。然而,单纯拥有长时间的药物半衰期,如果仅仅是因为药物在体内的分布容积过大,那么这样的半衰期并无实际的治疗意义。因为药物可能会过度分布在如脂肪组织等非靶标区域,导致真正需要治疗的部位药物浓度不足或完全缺乏。因此,确保药物能够准确、有效地作用于靶标,而不仅仅是在体内维持较长时间的存在,是药物研发过程中必须重点考虑的问题。
药物的有效性
有效性试验与功效试验在药物研发中各有侧重。有效性试验主要关注药物在真实临床环境中的治疗效果,这通常涉及到更为广泛和多样化的患者群体,以全面评估药物在不同条件下的实际疗效。而功效试验则更为专注于验证药物在特定条件下的作用机制,通常通过选择具有统一基线的狭窄患者人群来实现,以明确药物在特定条件下的作用效果。在评估新药物候选的成功潜力时,一个关键的挑战在于能否建立药物与治疗效果之间的直接联系。这种联系不仅需要药物与靶标的有效结合,还需要这种结合能够在实际临床环境中产生显著的治疗效果。以mGlu5受体PAM激动剂VU0092273为例,通过PET成像技术,研究人员能够观察到该分子在特定脑区域与受体的结合情况,进而证明了其在反向安非他命诱导的高活动行为中的治疗潜力。这一案例充分展示了靶标结合与治疗效果之间的直接关联,为药物研发提供了有力的支持。
药理学分析以更好地定义候选药物的功效
在药物开发过程中,药理学分析的重要性不言而喻。它不仅能够帮助定义候选药物的效能,而且通过特定功能测定来表征药物效果,为我们提供了药物在特定系统中功能的深入了解。这种分析不仅是对药物在单一系统功能的快照,更重要的是,它能够预测药物在复杂生物环境中的实际效果。药理学分析运用系统独立的参数,如亲和力和效力,这些参数能够应用于不同的药理模型,以预测药物在多种组织中的活性表现。这对于药物化学家来说至关重要,因为它为他们提供了优化候选药物设计和评估的依据。
在评估药物时,利用分子活性指标并避免复杂的系统行为是关键。例如,激动剂的效能虽然是一个古老而重要的指标,但它实际上是亲和力和效力两个分子特性的综合体现。通过深入分析这些参数,药物化学家可以更准确地了解药物与靶标的相互作用,进而预测药物在临床应用中的可能效果。
药理学在药物发现过程中的作用不容忽视。首先,它有助于我们深入了解疾病状态及所需,从而设计出能够针对特定疾病靶点的药物。其次,药理学分析能够帮助我们理解分子在临床设置中的复杂行为,为药物的安全性和有效性提供保障。最后,如果首选药物候选物失败,药理学分析还能为我们提供指导,帮助推进后续分子的研发,避免在失败的道路上重蹈覆辙。
在药物开发的实践中,药理学分析通过列举的实例,展示了在将体外活性转化为体内活性过程中可能遭遇的误解。这些例子包括慢性使用组织胺H2受体阻断剂后溃疡治疗效果的减失、-肾上腺受体激动剂未能提升血压、无法改善阿尔茨海默病中乙酰胆碱能神经递质下降的问题、GLP-1受体激动剂在糖尿病治疗中产生的副作用、CCR5内化剂在抗HIV-1感染中的失败尝试,以及偏向性血管紧张素受体调节剂在心力衰竭中的疗效不佳等。
这些实例突显了在药理学评估过程中全面了解候选药物化合物的重要性。特别是在将体外实验数据转化为体内疗效时,需要格外谨慎地进行分析和验证。这种谨慎的评估有助于避免在药物开发过程中走入误区,确保药物在临床应用中的安全性和有效性。
新药理学在药物发现中的应用
药理学在药物发现中扮演着至关重要的角色,它不仅能够识别和解决潜在的药物问题,更能够引入新的药理学概念,为药物发现开辟新的道路。最新的研究成果表明,高效性激动剂的高受体储备特性可能降低耐受性,而特异性针对某一信号通路的激动剂。如A1受体激动剂,能够实现有效治疗的同时减少不良反应。这些新见解不仅为药物研发提供了新的战略方向,也为我们提供了更有效的方法来优化药物候选物,推动药物发现的不断进步。
正向变构作用在药物发现中确实是一种有潜力的战略选择,特别是在针对特定受体如腺苷A1受体进行神经性疼痛治疗时。MIPS521作为一种腺苷A1受体的正向变构剂,通过稳定腺苷-受体-G蛋白复合物,成功地在大鼠神经性疼痛模型中实现了疾病特异性的镇痛效果。这一发现不仅为神经性疼痛的治疗提供了新的策略,也展示了正向变构作用在药物设计中的潜力。
此外,了解病理生理学对药物治疗进程的影响对于设计新药至关重要。以骨质疏松症的治疗为例,新数据揭示了PTH(甲状旁腺激素)或PTHrP(甲状旁腺激素相关肽)类似物与其受体的交互作用方式。这种交互作用能够不同程度地影响下游信号传导通路,特别是与甲状旁腺的分泌平衡状态相关的稳态作用。这些新见解为骨质疏松症以及甲状旁腺功能减退症的治疗提供了新的思路和方法。
近年来的药理学研究揭示了G蛋白偶联受体(GPCR)及其配体的新行为,这些新发现为治疗领域带来了新的可能性。首先,位置偏向性已经成为GPCR功能的重要特征,并在前导化合物的优化过程中发挥了关键作用。这种偏向性使得药物能够在特定的细胞位置产生效果,从而提高了治疗的精确性和有效性。传统上,受体内化被认为是信号终止的机制,但最近的研究表明,GPCR--arrestin复合物的胞内信号传导打破了这一观念。这一发现揭示了内化的受体可以继续从内质网发出信号,为细胞提供了一种新的响应机制。这种机制是受体和激动剂特异性的,意味着不同的受体和激动剂可能会产生不同的细胞响应。
以MC4受体为例,其天然激动剂-MSH仅在细胞表面产生效应,而MC4受体激动剂melanotan II则在内化后从细胞质内继续信号传导,这使其能够抵抗表面拮抗物AgRP的作用。这一发现为治疗与MC4受体相关的疾病提供了新的策略。此外,先前认为不对配体响应的GPCR,如CXCR7,也显示出可能产生相关细胞信号的特性。这一发现拓宽了我们对GPCR功能的认识,并可能开启新的治疗领域。在结构研究方面,高分辨率的结构解析为新药设计提供了重要依据。例如,PTH1R与Gs蛋白和小分子激动剂PCO371的高分辨率结构揭示了意想不到的结合方式,这为我们理解和设计针对B类GPCR的药物提供了重要线索。
GPCR变构配体效应的精准微调正逐步改变着我们对药物设计的理解,特别是在提高治疗靶向性方面展现出显著潜力。通过精细调整这些效应,我们能够实现对特定疾病靶点的精确打击,同时最小化对无关系统的干扰。其中,正性变构调节剂(PAM)的研究进展尤为引人注目,它们能够显著增强激动剂的效能,为那些传统上难以通过药物治疗的疾病提供了新的治疗策略。随着科研技术的不断进步,我们现在能够更深入地研究PAM与GPCR之间的相互作用机制,以及它们如何影响激动剂的活性和选择性。这些新见解不仅为药物设计提供了新的工具和方法,也为治疗多种疾病提供了更多的可能性。
总之,GPCR变构配体效应的微调为我们打开了一个全新的药物设计领域,让我们能够更精准地针对特定疾病靶点进行治疗。随着科研工作的深入,相信未来我们将看到更多基于这一策略的创新药物问世,为患者带来更多的希望。
总结
•
新药物发现和开发是一个综合而复杂的过程。首先,科学家们识别与疾病相关的靶点,如G蛋白偶联受体(GPCRs),并深入理解其功能和行为。随后,通过配体筛选,他们寻找能与这些靶点有效互动的潜在药物分子。在这一过程中,对GPCRs等复杂靶点的深入了解对于候选药物的预测和决策至关重要。科学家们借助先进的实验技术和分析工具,如高通量筛选和计算模拟,来详细研究这些靶点的行为,以便更准确地预测候选药物的药理特性。在获得候选药物后,科学家们还需通过体外和体内实验,全面评估其药效、药代动力学、安全性和耐受性等方面。这些评估结果不仅指导药物的进一步优化,还为后续的临床试验提供了重要依据。随着技术的进步,新药物发现和开发的效率与成功率不断提高,为治疗各种疾病提供了更多可能。我们期待未来能有更多安全、有效的药物问世,为患者带来福音。
参考文献
[1] Kenakin, T. Know your molecule: pharmacological characterization of drug candidates to enhance efficacy and reduce late-stage attrition. Nat Rev Drug Discov (2024). https://doi.org/10.1038/s41573-024-00958-9
PROFILE
Terry Kenakin
北卡罗来纳大学医学院药理学

Terry Kenakin博士是北卡罗来纳大学医学院药理学的杰出教授,他在药理学领域享有崇高的声誉。他的职业生涯丰富多彩,曾在Burroughs-Wellcome公司从事药物发现工作长达7年,之后在GlaxoSmithKline(葛兰素史克)公司担任重要职务长达25年。在这段漫长的职业生涯中,他积累了丰富的药物研发经验,为药理学领域的发展做出了巨大贡献。
Kenakin博士不仅在实践领域成就卓越,在学术领域也颇有建树。他是多本药理学领域书籍的作者,这些书籍为后来的研究者提供了宝贵的学术资源。此外,他还担任《Journal of Receptors and Signal Transduction》的主编,并在多个编辑委员会任职,为学术交流和传播做出了重要贡献。
Kenakin博士的杰出成就得到了国际社会的广泛认可。他因在基础和临床药理学及毒理学领域的卓越贡献,荣获了多个国际奖项。其中包括2008年挪威药理学会颁发的Poulsson药理学奖、2011年荷兰药理学会颁发的Ariens奖以及2014年英国药理学会颁发的Gaddum纪念奖。
END
文案 | 晶体转业
排版 | 晶体转业
审核 | 晶体转业
发布|姜笑南
世界生命科学大会
RECRUIT
关注我们,获取生命科学
学界前沿|促进更多的学术交流与合作
业界前沿|促进更快的产品创新与应用
政策前沿|促进更好的治理实践与发展
本文作者可以追加内容哦 !

