转自:
允咨GMP制药技术
2022-05-11 08:57河北石家庄允咨医药科技有限公司
Pre-IND+IND+NDA(全流程图)
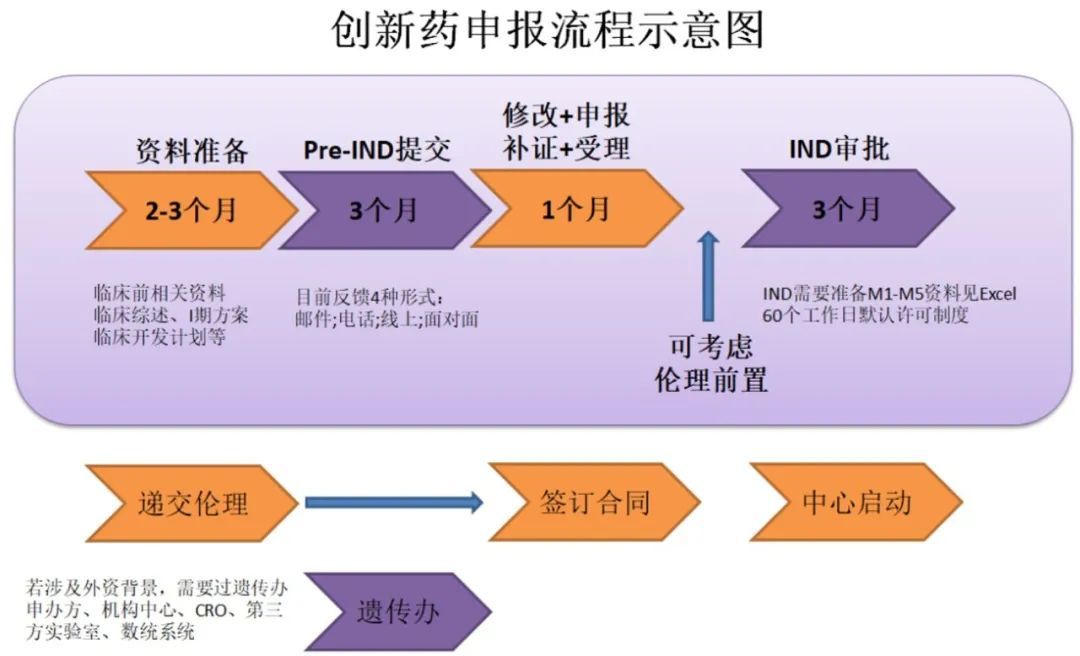

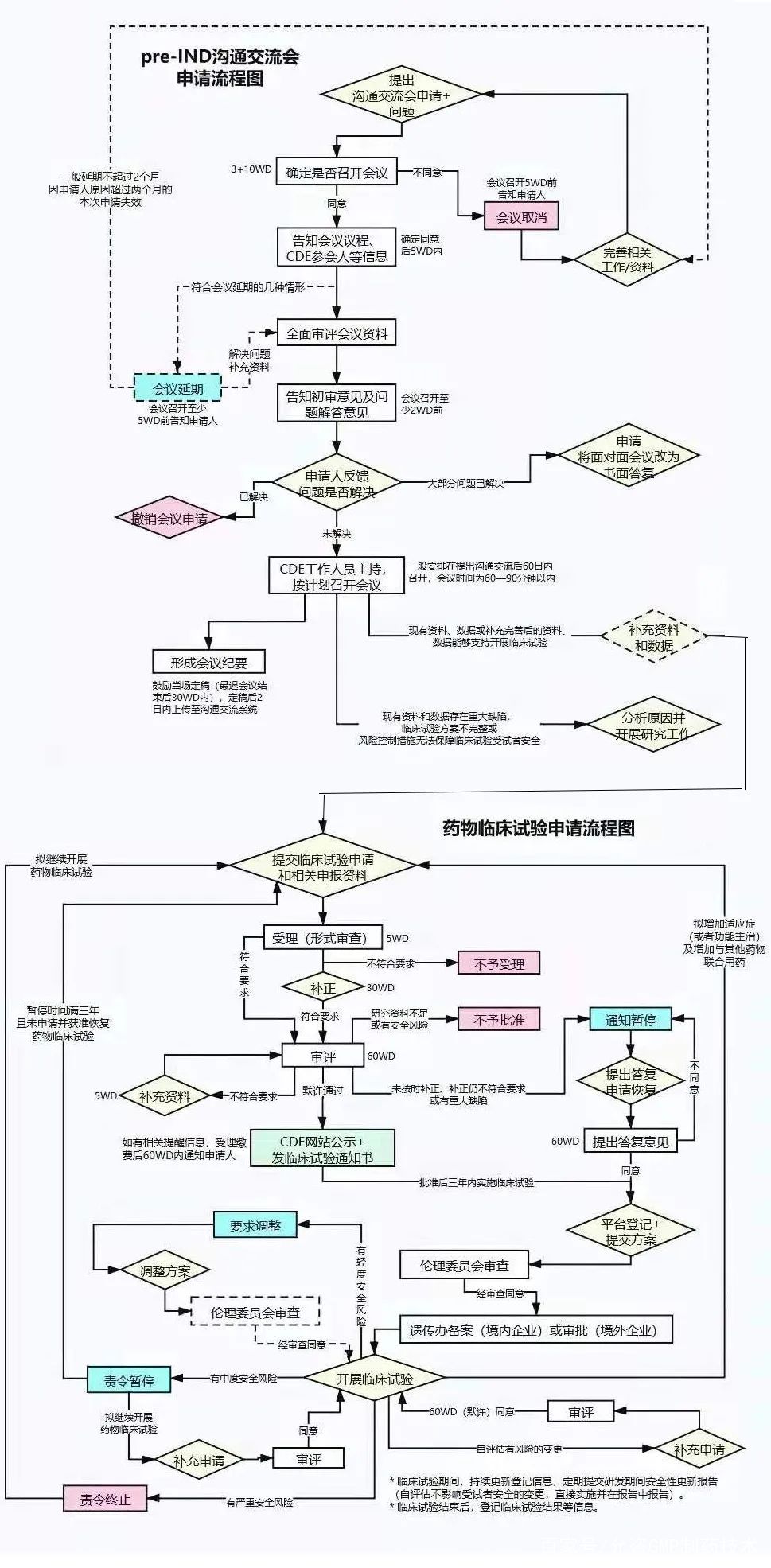
药物开发从药物发现和筛选开始,企业需开展一系列的合成工艺、制剂工艺、质量研究等等前期工作,且还需开展一系列动物毒理、药效和(或)药代动力学研究,得到药物安全性方面的初步证明后,方可申请临床试验。
02 Pre-IND会议
向FDA申报IND,首先,企业需要有在美办事处或者寻求合适的美国代理人,负责与FDA的沟通联络。
FDA建立了多种有效的和企业的沟通机制,比如各种与企业的正式会议,如Pre-IND会议、二期临床结束后会议、NDA/BLA递交前会议等等。通过会议,企业可以获取更多FDA对于药物开发方面的建议、清楚FDA的要求,减少走弯路、走错路的可能性,提高申报成功率。顾名思义,在申报IND之前企业与FDA的会议,即称为Pre-IND会议。
为此,企业需要准备一个Pre-IND package,预先告知FDA药物的基本信息、研究现状、初步的研究计划等等,通常包括CMC、临床前药理毒理、临床试验方案大纲、已有人体临床经验(如有)的综述资料,而最重要的内容当数企业拟在Pre-IND会议上与FDA讨论的问题。准备问什么样的问题可以说是整个Pre-IND阶段最重要的一项工作。
一般说来,企业需在计划和FDA开会前60天左右,向FDA提出会议申请。FDA在收到会议申请后,一般在14天内作出会议安排。之后,企业向FDA递交Pre-IND简报文件(一般在会议前4周左右递交)。FDA通常会回复对拟讨论问题的初步意见,企业可以了解到FDA对问题的初步看法。通常企业只有一次与FDA开Pre-IND会议的机会,其重要性不容小觑。
在Pre-IND会议结束后数个工作日,FDA会提供正式的Pre-IND会议纪要,记录FDA和申报者对于问题的意见和讨论结果。申报者也可以在会后提供自己的会议记录给FDA,表达自己对讨论情况的理解,避免双方理解上的误差。
03 IND申报文件包
根据Pre-IND会议与FDA的讨论结果,申报者进行IND申报文件包的编写。
IND申报文件包主要包括9部分的内容:首页函、FDA 1571表;目录;引言和总体研究计划;研究员手册;临床研究方案;化学、生产和质量控制信息;药理和毒理信息;已有人体临床经验;额外信息;需要注意的是,在IND申报文件包中,还需提交相关原始完整研究报告,如毒理研究报告等。
04 申报IND审评
审评工作从FDA收到IND申报文件包开始计时,FDA需在30天内完成审评。在审评后期,FDA可能会和申报者就某些问题开展讨论。审评结果一般包括三种情况:允许开始临床;部分临床限制以及临床限制。
IND申报的流程比较复杂,在整个的IND过程中每一步都存在着失败的可能。有报道称IND总体失败率超过了90%。导致药物研发申报失败的前3位因素分别是药代动力学、临床有效性和毒性研究。
05 药品上市后许可申请
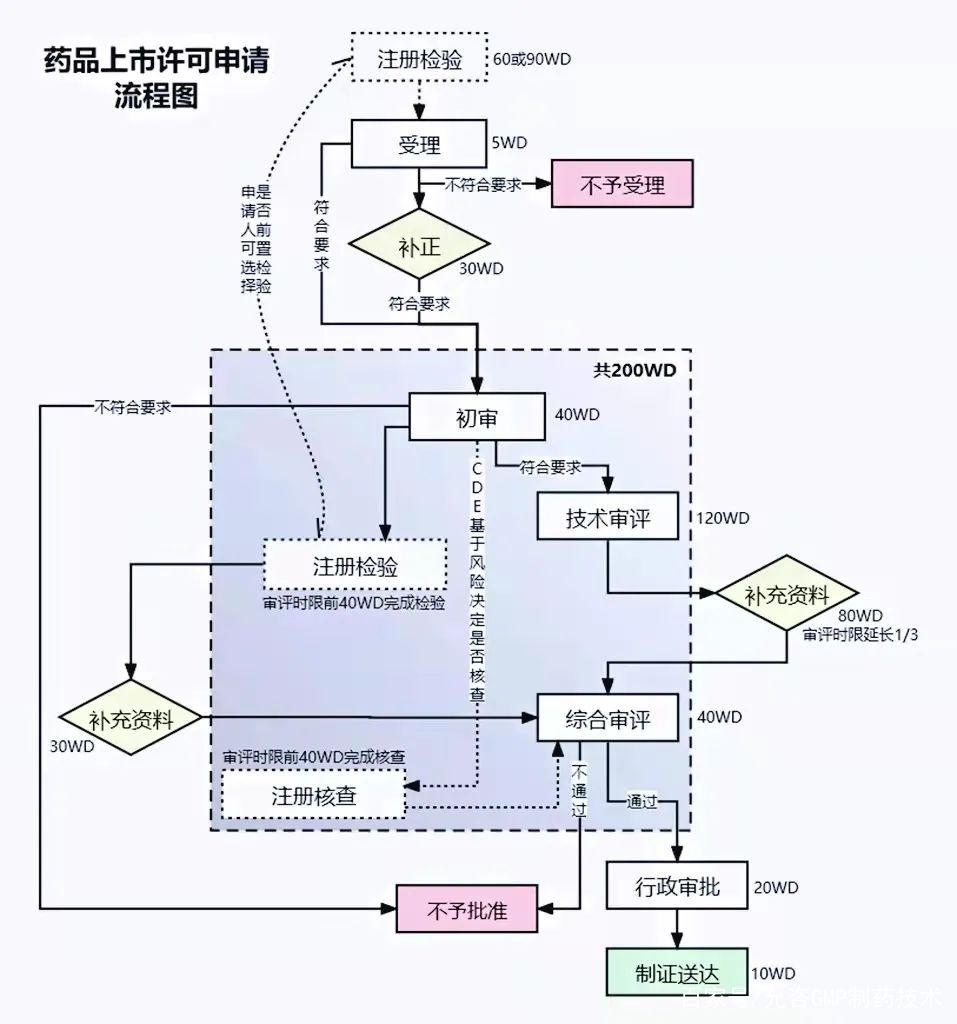
到了第二大步骤,离成功上市越来越近![[上涨]](http://gbfek.dfcfw.com/face/emot_default_28x28/emot78.png "上涨")
本文作者可以追加内容哦 !

