8月20日,再生元宣布已收到FDA就其BCMA/CD3双抗Linvoseltamab的BLA申请发出的CRL,指出因化学、生产和控制(CMC)问题,该药物的上市申请未能获得批准。
这是继FDA拒绝批准odronextamab之后,又一款关键的TCE双抗的BLA被FDA拒绝。
再生元表示,此次申请被拒的原因是第三方灌装与封口制造商所生产的另一家公司的上市候选产品在批准前的检查结果未通过,从而产生了连带效应。不过,该第三方制造商已通知再生元,他们认为相关问题已得到解决,其设施目前正等待FDA的重新检查,预计这一检查将在未来几个月内进行。该公司指出,FDA没有对linvoseltamab的安全性和有效性提出问题。
Linvoseltamab(REGN5458)是靶向BCMAxCD3 的双特异性抗体,旨在将多发性骨髓瘤细胞上的 B 细胞成熟抗原 (BCMA) 与表达 CD3 的 T 细胞桥接,以促进 T 细胞活化和癌细胞杀伤。再生元已在早期阶段启动linvoseltamab的3期临床,并在今年2月份FDA提交BLA。此前,FDA已授予linvoseltamab治疗多发性骨髓瘤的快速通道资格。
BLA主要基于I/II期linker-MM1试验的数据支持,这是一项关键的剂量递增和剂量扩展试验,招募了280多名患者。评估linvoseltamab在R/R MM患者中的作用。282例入组患者都接受过至少三线治疗或出现三重耐药。
该临床试验(n=117)中接受剂量为200mg的linvoseltamab治疗的患者中观察到客观缓解率为71%,46%达到完全缓解或更佳。然而,在安全性上,去年12月的数据显示,接受 200 毫克剂量双特异性抗体治疗的所有 117 名患者都发生了不良事件,其中 85% 的严重程度为 3 级或更严重。14名患者死于治疗中出现的不良事件。
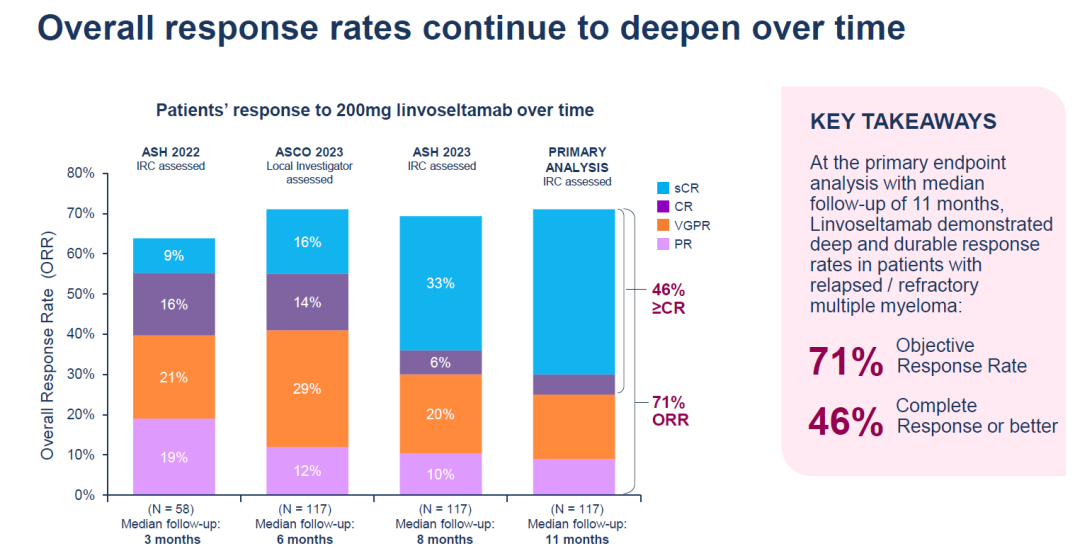
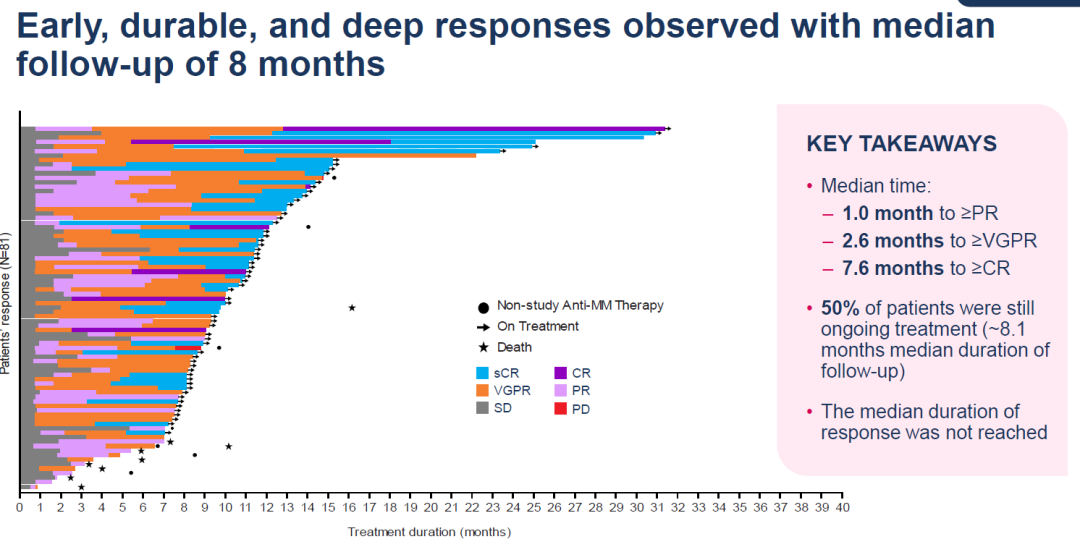
今 年 6 月,再生元发布了 LINKER-MM1 的更新数据,该数据验证了 linvoseltamab 的高缓解率,并显示出良好的总体生存期和无进展生存期。

除LINKER-MM1外,再生元还在进行III期验证性研究LINKER-MM3,该研究旨在评估linvoseltamab在经过大量预处理的复发或难治性多发性骨髓瘤患者中的临床益处。根据其 clinicaltrials.gov 页面,LINKER-MM3预计将于2032年完成。
小结
Regeneron今年不太走运,先是CD20/CD3双抗odronextamab被拒绝,现在又因为生产问题导致BCMA/CD3双抗被拒绝。
无论出于何种原因,Regeneron现在在BCMA和CD20 双抗已经在竞赛中都已落后。此前,FDA已经批准了两种BCMAxCD3双特异性药物:强生的Tecvayli和辉瑞的Elrexfio。
本文作者可以追加内容哦 !

