越来越多的出海成功案例传递出的积极信号是,面对FDA这个依旧稍显陌生的市场,中国药企已然能够走得越来越稳妥。
2023年下半年,中国创新药行业异常火热,一扫年初多款创新药出海失利给行业带来的阴霾。国产创新药出海提速,君实生物的特瑞普利单抗、和黄医药的呋喹替尼和亿帆医药控股子公司亿一生物的艾贝格司亭注射液等成功闯关,陆续获得美国食品药品监管局(FDA)批准上市。
获批后的仅仅48小时内,呋喹替尼已经开出在美国市场的第一张处方。之后的一周,呋喹替尼就进入了全世界最权威的治疗指南之一的NCCN(美国国家综合癌症网络)指南。这一天,和黄医药的公关团队集体在朋友圈“刷屏”了,但是没有人觉得一天数十条的朋友圈有多么“出格”。
作为我国首个自主研发生产的抗癌新药,从在美国正式成立临床和注册团队开始,这一天,和黄医药等了5年。
对于国内医药行业来说,冲向FDA是一种相互竞争但也互为依仗的存在。在竞争激烈的创新药环境中,每个药企都渴望成为那个能够撞开第一大医药市场大门的人,也都在为“伙伴”的成功欢呼喝彩。
原因无他,成功走进美国市场,代表的不仅仅是一款产品的获批,而是得到了这个全球门槛最高市场的盖章认证,这是对中国创新能力整体的认可。
FDA这块金字招牌的价值,大概是在2000年前后才被国内逐渐认识到。2001年,中国加入WTO。站在世界舞台上的中国制药界发现,自己并没有多少话语权。冲击FDA,在彼时开始,成了企业和行业的目标和“心病”。把药卖到美国去,本身在经济效益上就是一件很可观的事情。更何况获得FDA批准之后,对打入欧盟、日本等发达国家地区市场有很大帮助。但自2000年后的20多年间,国内制药业技术实力普遍较弱,研发创新能力几乎为零,本土只有仿制没有研发,能达到FDA标准的品种屈指可数。
某种程度上,越来越多的出海成功案例传递出的积极信号是,面对一个全然陌生的市场,中国药企已然能够走得越来越稳妥。这也意味着,中国原研创新药的出海大潮正加速来袭。
谁是下一个闯关者?
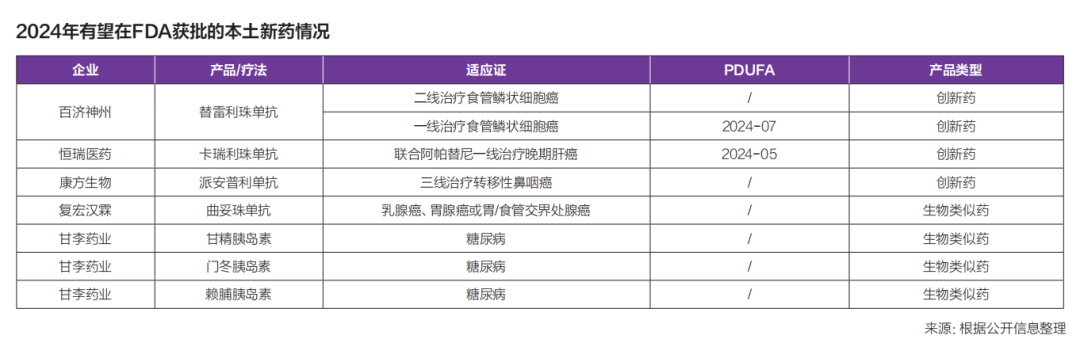
在一年5款产品的辉煌下,行业的期待值被拉升。接下来,还能否“复制”这一速度?谁又会成为2024年第一个闯关成功的选手?
根据公开信息来看,共有8款产品/适应证在FDA的待审评过程中,其中4原创新药全部为单抗。
百济神州一向以适应证打天下,仅泽布替尼在FDA获批的适应证就达到了5个。明年,百济神州在FDA的“场子”可能要交给替雷利珠单抗。此前,替雷利珠单抗已有12项适应证获得NMPA批准,是目前在中国获批适应证数量最多的PD-1抑制剂。
2023年7月,FDA完成了对替雷利珠单抗的生产检查。这也意味着,这款曾经与诺华分分合合的重磅产品,终于把冲向美国市场的日程,拉回了正轨。
在“原创”之外,另外一个赛道已经悄悄浮出水面。12月7日,百奥泰宣布,其在研贝伐珠单抗生物类似药Avzivi获美国 FDA批准上市。Avzivi是百奥泰第二个获得美国 FDA上市批准的产品,也是第二个由中国药企研发且获得 FDA上市批准的生物类似药产品。
值得关注的是,在2024年有可能冲向FDA的8款产品/适应证中,有4款为生物类似药。复宏汉霖的曲妥珠单抗、甘李药业甘精胰岛素、门冬胰岛素、赖脯胰岛素正在FDA上市审评中。
其中,最受关注的无疑是复宏汉霖的曲妥珠单抗生物类似药汉曲优,此前汉曲优分别在欧洲和中国双上市,是首个中欧双批的国产单抗生物类似药,创下中国Biosimilar的历史。目前汉曲优已经在全球41个国家上市,成为国产生物类似药出海的标杆。
据医药魔方数据库不完全统计,国内目前共有生物类似药管线450条。除去管线状态不活跃和状态未知的项目,共有活跃管线393条(含国内企业引进管线),占到全球生物类似药活跃管线的47%。从数量上看,近半数的生物类似药管线都在中国,处于领先地位。生物类似药的规模和实力,或许决定了它未来将是冲向FDA的又一大主力。随着各个原研药在欧美的专利过期,生物类似药似乎也成了各家必争之地。
不过,与从2000年前后便开始探索的创新药出海相比,在政策上,我国在2015年发布《生物类似药研发与评价技术指导原则(试行)》就落后了一步,而第一个单抗生物类似药2019年才上市。
另外一个值得深思的问题是,我们的生物药类似药赴美上市还要面临重重阻力。FDA对生物药的审评审批有着很高的临床以及生产标准,尤其是同类型药物已经“珠玉在前”的情况下审批更为审慎。事实上,去年信达生物终止了与Coherus关于贝伐珠单抗类似药的合作,收回其在北美的权益,信达当天的股价就下跌了8%。信达给出的原因是“评估贝伐珠单抗生物类似药北美市场动态以及受疫情持续影响开发延误”。疫情的影响终归是暂时的,但北美生物类似药市场激烈的竞争恐怕是“市场动态”这四个字背后的含义。
欧美发达国家市场的确诱人,但想成功打入并不简单,极高的多维度审评标准(临床、生产、同类药对照)、飞速变化的竞争格局无一不成为国产生物类似药闯关的“拦路虎”。毕竟很难有企业能像复宏汉霖一样占尽天时地利人和。
为什么是他们?
在想要回答“还有谁”这个问题之外,或许有另一个问题的答案更值得深究:为什么是他们?
呋喹替尼早在2020年便获FDA授予“快速通道”资格,并于同年启动了全球Ⅲ期多中心临床试验FRESCO-2研究。虽然正值疫情高峰,FRESCO-2研究快速地完成了患者招募。在呋喹替尼获批之前,美国三线结直肠癌市场是瑞戈非尼和TAS-102二分天下的局面。这两款药物分别于2012年和2015年获FDA批准用于肠癌治疗。而在靶向疗法方面,超过十年时间里,转移性结直肠癌领域一直未有新药出现,直到呋喹替尼的获批改变了这一僵局。结直肠癌作为一个主流癌种,要挑战现有市场,说来并非轻巧。和黄医药拿出的关键砝码是其卓越的疗效,并凭此快速进入NCCN指南。
获批一周后,呋喹替尼便进入了全球肿瘤医生的宝典—NCCN指南。最新版的NCCN指南已将呋喹替尼纳入三线推荐方案,而对于一线治疗时已通知接受过奥沙利铂及伊立替康的患者,更将呋喹替尼推荐为二线方案。
在呋喹替尼设计之初,和黄医药的理念就是通过提高激酶选择性,尽可能降低脱靶毒性和提高耐受性,使其在拥有理想药效的同时具备良好的安全性。
从呋喹替尼的经验来看,如今,随着先行者们的逐步探索,出海答卷已经日渐清晰:过硬的产品质量、世界级研究数据、全球布局的临床注册能力。
其中,过硬的产品质量是根本。此前,泽布替尼在全球性Ⅲ期头对头研究中,在安全性、优效性等多个关键评估方面,均优于伊布替尼。这项头对头研究曾被百济神州联合创始人、董事长兼 CEO欧雷强称之为公司历史上最重要的临床数据之一。这也使得泽布替尼备受认可成为Best-in-class。而首个出海的PD-1替雷利珠单抗也具有差异化竞争优势,其分子设计旨在最大限度地减少与巨噬细胞中的 Fc受体结合,具有显著增强抗肿瘤活性的特点。
世界级研究数据则是必选项。这要求药企从立项一开始就高标准、严要求,以国际化视角去设计实验方案并贯彻执行。全球临床注册能力,决定着出海的效率与进度。国际多中心临床入组地点较多,对药企的研发团队及运营团队有着极高的要求,不仅要严格把握国际准则、入组患者的入排标准、试验质量控制,还要非常熟悉海外临床注册法规的各项要求。
监管通关的下一步
从泽布替尼、西达基奥仑赛,再到如今的特瑞普利单抗、呋喹替尼,一个个创新产品的成功突围,意味着中国原研创新药开始与世界接轨。不过,严格来说,当前还只是出海的上半场。
但此前,PD-1在国内市场的定价,中国药企的原研药价格均不足K药、O药的50%,曾经让国产新药被贴上了“低价”的标签。
价格,是中国创新药冲向海外市场不可或缺的拼图。一年5款产品的成功闯关,论证了中国药企能够做出兼具品质、创新的好药;然而,摘掉“低价”标签,才是中国药企海外定价和商业化的最终一步。
11月27日,君实刚获批不久的特瑞普利单抗在美国的批发采购价曝光,为每瓶8892.03美元,约合人民币62982元。在中国,同规格药物的售价为1912.96元,因此,特瑞普利单抗在美国的价格是中国价格的33倍。
随后,呋喹替尼的价格也正式公布。1mg和5mg规格的产品,单盒售价分别为6300美元和25200美元。而在国内,呋喹替尼的最新医保定价为1885.38元每盒(1mg*21粒),2513.70元每盒(5mg*7粒)。也就是说,呋喹替尼在美国的价格是中国价格的24倍。
这两款国产新药在美国的价格,表现了国际社会对中国生物医药新产品的认可,意味着我国生物医药行业已接近国际先进水平。
1月22日,中共中央办公厅、国务院办公厅联合印发了《浦东新区综合改革试点实施方案(2023-2027年)》(以下简称“《方案》”)。
《方案》提出,要完善开放合作的国际协同创新机制。探索制定人工智能、生物医药等全球重大前沿科技领域伦理规则,建立协同审查机制,构建伦理审查快速通道。创新药在国内的定价,也关乎到中国创新药在全球价值的最大化,尤其当中国的创新药开始规模化出海、接二连三闯关FDA成功之后。中国的医药研发产业目前正在经历资本寒冬,较低的创新药价格非常不利于吸引投资。在这一背景下,出台这一政策,表现政府对创新药行业发展的巨大支持。从更高层面说,这也是目前我国完善科技创新体系、建设开放创新生态的重要举措。
不过,如何结合不同市场的医保政策以及产品的创新性、差异化来确定产品的创新价值,进而确定产品在未来市场的生命周期以及定价是国内创新药企必须做的功课。药企出海的意义不仅仅是与全球接轨,也是整个行业真正形成良性的创新循环、惠及更多患者的关键,这也是我国生物医药产业实现“从大到强”的必经之路。
本文作者可以追加内容哦 !

